![澳门威斯人游戏平台[亚洲]全站最新版V11.37.7](/wp-content/themes/2023/images/banner/technical-resources.jpg?v=18)
摘要:本文主要从基础原理与实验设计思路两个方面讲述了蛋白纯化实验中离子交换色谱的应用,并附有AKTA离子交换色谱操作案例。
离子交换色谱是蛋白纯化技术中常用的一种纯化方法,其原理是指被分离物质所带的电荷可与离子交换剂所带的相反电荷结合,这种带电分子与固定相之间的结合作用是可逆的,在改变pH或者用逐渐增加离子强度的缓冲液洗脱时,离子交换剂上结合的物质可与洗脱液中的离子发生交换而被洗脱到溶液中。由于不同物质的电荷不同,其与离子交换剂的结合能力也不同,所以被洗脱到溶液中的顺序也不同,从而被分离出来。
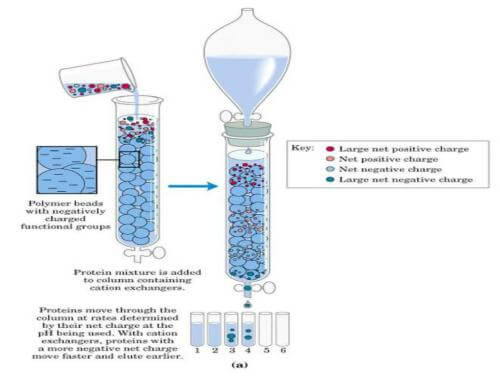
离子交换剂是由不溶于水的网状结构高分子聚合物骨架构成,骨架上有许多共价结合的带电基团,如果侧链是带正电基团,就可与带负离子相结合,称为阴离子交换剂,吸附带负电蛋白质。如果侧链是带负电的基团,则称为阳离子交换剂。强离子交换树脂在宽pH范围内保持离子化,而弱离子交换树脂只在窄pH值内离子化。
| 类型 | 名称 | 英文符号 |
|---|---|---|
| 阴离子交换剂 | 二乙基氨乙基 季氨基乙基 季氨基 三乙基氨乙基 氨乙基 |
DEAE QAE Q TEAE AE |
| 阳离子交换剂 | 羧甲基 磺丙基 磺甲基 磷酸基 |
CM SP S P |
绝大多数重组蛋白纯化都要用到离子交换。离子交换色谱的基础是高分辨率,可以直接放大规模应用在工业上,柱再生容易,还可以使蛋白浓缩。大多数蛋白质的静电荷是负值,因此阴离子交换色谱的应用最为广泛。
离子交换介质首先要考虑目的分子的大小,因为目的分子会影响其接近介质上的带电功能集团,因此也会影响介质对目的分子的动力载量,从而影响其分离。
对于大多数纯化步骤来说,建议从开始的阶段使用强离子交换柱,可在摸索方法的过程中有一个宽的pH范围。对于已知等电点的蛋白质,可根据其等电点来选择。而未知等电点的蛋白质,在实际操作中常采用这样的方法,先选择一个阴离子交换剂,再选择一个中性的pH缓冲液,将蛋白质样品透析至pH7.0,然后过阴离子交换柱。根据过柱后的结果确定下一个使用的缓冲液pH。
离子交换色谱的流动相必需是有一定离子强度的并对pH有一定缓冲能力的溶液。为了避免目的蛋白失活,使用缓冲液可稳定流动相的pH,使之在色谱过程中不发生明显变化,同时可稳定目的分子上的电荷量,保证色谱结果的重要性。
选择缓冲液一般按照以下原则:阳离子交换剂应选用阴离子缓冲液,可用柠檬酸盐、磷酸盐、醋酸盐、甘氨酸盐等;阴离子交换剂应选用阳离子缓冲液,可用烷基胺、Tris、氨基乙醇胺、乙二胺、咪唑等;起始缓冲液的浓度应尽可能低(<100mmol/L)这样可以使色谱柱上更多的吸附分离物质;缓冲液应不含会影响被分离物质活性和溶解度成分,洗脱时尽量不采用pH梯度洗脱。
离子交换色谱通常选用粗短柱,即高径比小的色谱柱。典型的离子交换柱高度在5~20cm,高径比一般小于5。如果需要增加离子交换剂的体积,只能从增加柱的直径而不能增加其高度。如果是连续梯度洗脱,可以适当增加柱的长度。
离子交换是蛋白纯化中的重要手段,即可以用于捕获阶段,也可以用于精纯阶段。
20mmol/L磷酸盐缓冲液,pH6.5,用10柱体积平衡缓冲液平衡柱子,直至pH及电导率监控显示与平衡缓冲液的pH及电导一致。
加样
根据柱体积及装柱的最大流速确定上样流速,上样流速不超过装柱最大流速的75%,同时,上样流速也要尽可能低,保证样品和介质充分发生作用。
洗脱
上样结束后,用平衡缓冲液冲洗1~2柱体积,使UV280响应信号重新回到基线。
之后用含有0.5mol/L NaCL的溶液洗脱,冲洗2~3柱体积,收集色谱峰。






澳门威斯人游戏平台[亚洲]全站最新版V11.37.7 Nanjing Detai Bioengineering Co.,Ltd. ©2024 All Rights Reserved
苏ICP备2021019379号-1 | 网站地图 | 用户协议 | 隐私政策